Diagnosis: adult onset acid maltase deficiency (Pompe disease).
The next step was testing for alpha acid glucosidase (GAA) enzyme activity in the whole blood: Acid alpha-glucosidase was < 2.5 pmol/punch/h (Normal >7.4)
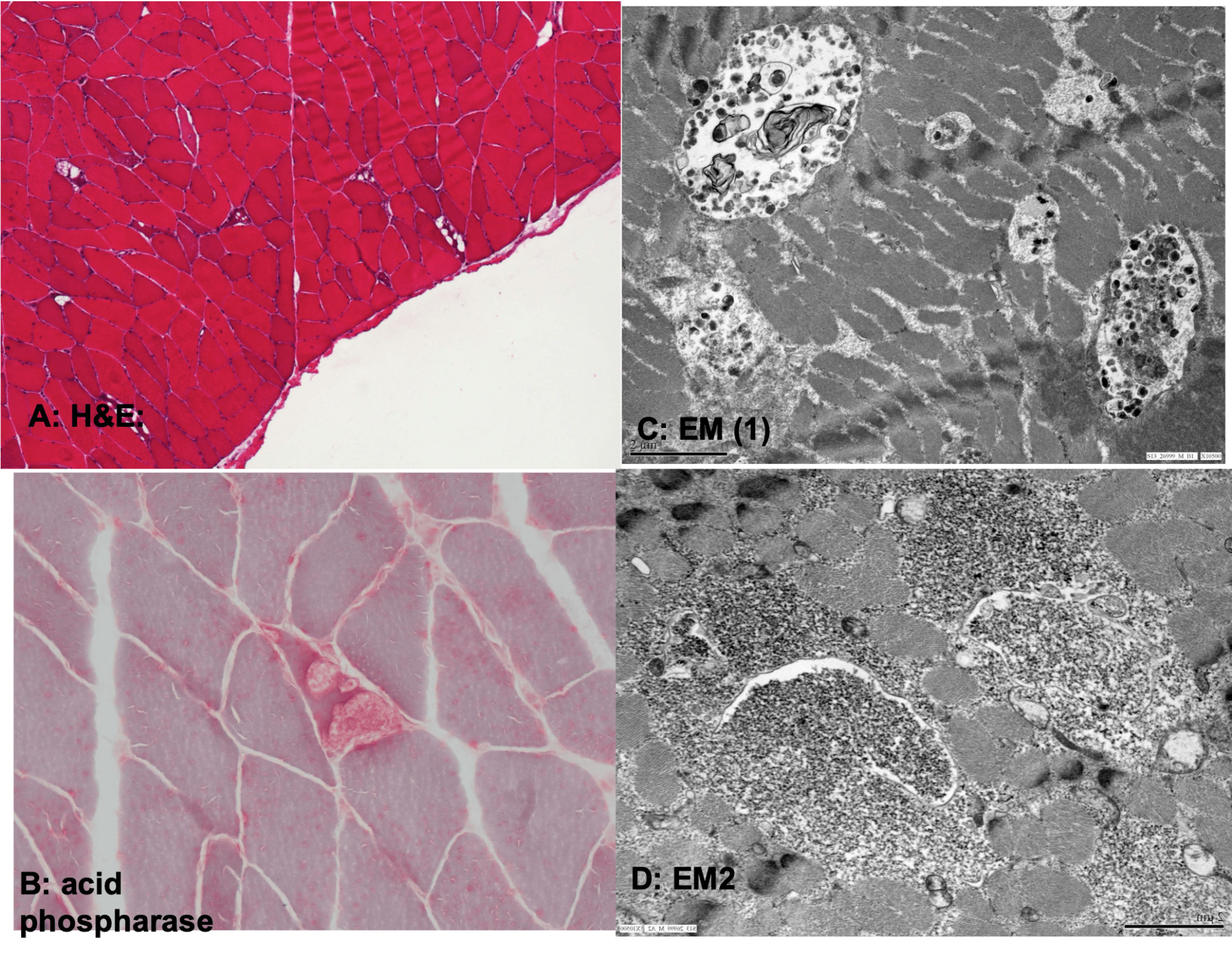
The patient also had undergone a muscle biopsy prior to the GAA enzymatic assessment. H&E staining (A) showed increased variation of myofiber size, random admixture of atrophic myofibers, and occasional myofibers with vacuolar changes. The vacuoles corresponded to increased cytoplasmic glycogen and stained positively with acid phosphatase stain (B). EM showed increased cytoplasmic pools of glycogen which was at least partly membrane bound (C, D).
Pompe disease (aka glycogen storage disease type 2) is a rare, autosomal recessive disease which is caused by mutations in GAA (mutation in long arm of chromosome 17). Age of onset and severity of Pompe disease vary depending on the residual GAA enzymatic activity, itself a function of the type of mutation. Infantile Pompe disease (residual enzymatic activity <1%) presents with general weakness (floppy baby syndrome), cardiomyopathy , progressive respiratory failure and death before 1 year if untreated. Pompe disease may also manifest in children and adults, in which case a myopathy is the cardinal feature. Pelvic girdle and truncal muscles are commonly affected and scapular winging, weakness of neck flexors, and camptocormia could be significant. Respiratory muscle weakness is the result of truncal and diaphragmatic weakness. Cranial muscles are not affected although ptosis has been reported. Cardiomyopathy is not a feature of adult onset Pompe disease.
Measurement of GAA enzyme activity (in dried blood spot, purified lymphocytes, cultured fibroblasts) should be considered as a screening test even before a muscle biopsy in limb-girdle syndromes with significant diaphragmatic or truncal weakness. If significant decrease of GAA is found, a second test (GAA measurement is purified lymphocytes, muscle or fibroblast culture , genetic testing) should be done to confirm the diagnosis. CK is normal to x10 times elevated in adult onset Pompe disease. It should be noted that muscle biopsy may not show vacuolar myopathy and glycogen accumulation if the biopsy is not performed on the right muscle (i.e. false negatives could occur).
It should be emphasized that early diagnosis of Pompe disease is essential, as it could result in improved outcome with enzyme replacement treatment (ERT). It is recommended that symptomatic patients be started on ERT and be re-evaluated one year later to determine whether the treatment should continue. Presymptomatic patients are to be closely observed and be treated when the symptms start.
Suggested readings:
1. Vissing J, Lukacs Z, Straub V. Diagnosis of Pompe disease: muscle biopsy vs blood-based assays. JAMA neurology. 2013;70(7):923-7.
2. American Association of N, Electrodiagnostic M. Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle & nerve. 2009;40(1):149-60.
3. Cupler EJ, Berger KI, Leshner RT, Wolfe GI, Han JJ, Barohn RJ, et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle & nerve. 2012;45(3):319-33.